Scaling Imputation with a 150,000-Sample UK Biobank Reference Panel Using GLIMPSE2
The UK Biobank's WGS release created an unusually large reference panel — large enough to strain conventional imputation pipelines. Here's how GLIMPSE2 was redesigned to handle it efficiently.
The UK Biobank’s release of whole-genome sequences from over 150,000 individuals made a reference panel of unprecedented size available to researchers. That brought an immediate practical question: how do you actually use a panel that large for imputation, when most existing tools were designed for panels an order of magnitude smaller?
This post describes the core ideas behind GLIMPSE2 and the design choices that allowed it to handle this scale.
The problem with scaling
Standard low-coverage imputation methods — including our own GLIMPSE1 — work by first selecting a subset of reference haplotypes for each target individual (typically via the Positional Burrows-Wheeler Transform), then running a Hidden Markov Model (HMM) over that subset. For moderate panel sizes this works well, but computation grows roughly linearly with the number of reference haplotypes.
At 280,000 haplotypes, that growth becomes a bottleneck. Running GLIMPSE1 unmodified on the full UK Biobank reference would have been impractical at biobank scale without a substantial change to the algorithm.
The GLIMPSE2 solution: split-and-merge
The central idea in GLIMPSE2 is straightforward: rather than running a single HMM over all selected haplotypes, we split the reference into smaller, overlapping subsets, run the HMM independently on each, and then combine the resulting posterior probabilities. This has several practical consequences:
- Sublinear scaling: As the panel grows, we can include more haplotypes in aggregate without requiring each individual run to be proportionally larger. Compute cost grows slowly relative to panel size.
- Parallelism: Each subset can be processed independently, which maps well onto HPC environments.
- Retention of rare variant accuracy: By ensuring each subset contains locally relevant haplotypes, the method maintains reasonable accuracy even at very low minor allele frequencies.
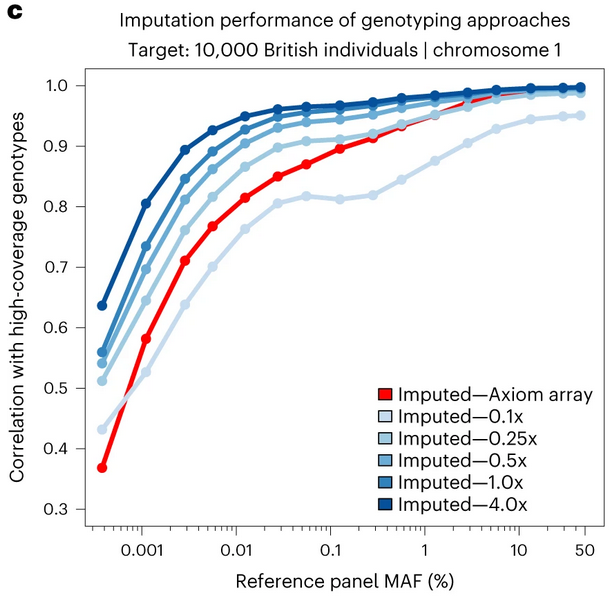
Figure 1: Imputation performance comparison across the frequency spectrum at different sequencing depths. GLIMPSE2 maintains useful correlation with high-coverage genotypes down to 0.1x coverage, though accuracy at the lowest depths and rarest variants should be interpreted with care.
What we achieved
Using GLIMPSE2 with the 150,000-sample UK Biobank WGS reference panel, we were able to impute target samples at a cost of less than $0.1 per sample with cloud pricing at the time. The resulting dataset covered approximately 580 million markers per individual.

Figure 2: GWAS power across 22 UK Biobank phenotypes. Imputed low-coverage WGS at 0.5x and above compares favourably with SNP array genotyping (Axiom), particularly for rarer variants not well captured by arrays.
Broader context
Low-coverage WGS followed by imputation has become a credible alternative to SNP arrays for large-scale studies, with per-sample sequencing costs continuing to fall. Arrays have the advantage of mature workflows and predictable costs, but they are limited to pre-defined marker sets and may not capture population-specific variation well.
The suitability of lcWGS + imputation depends on study design, target variant classes, and available reference panels, but the feasibility of running it on very large cohorts is now well established.
Full paper: Rubinacci et al., Nature Genetics 2023. doi:10.1038/s41588-023-01438-3